Update: I have tried to automatize the open source ab-initio codes Quantum Espresso and YAMBO to compute ground state and excited state energies. These automatic scripts using Quantum Espresso (6.7MAX) +YAMBO (v5.1) are now available in my GitHub I have tried to write the flow in a super easy to understand format, so that anyone can run these lengthy and cascaded consecutive flows without losing much time in between. The folder names are compatible also to the yambopy process-flow.
Electronic Structure Theory Group
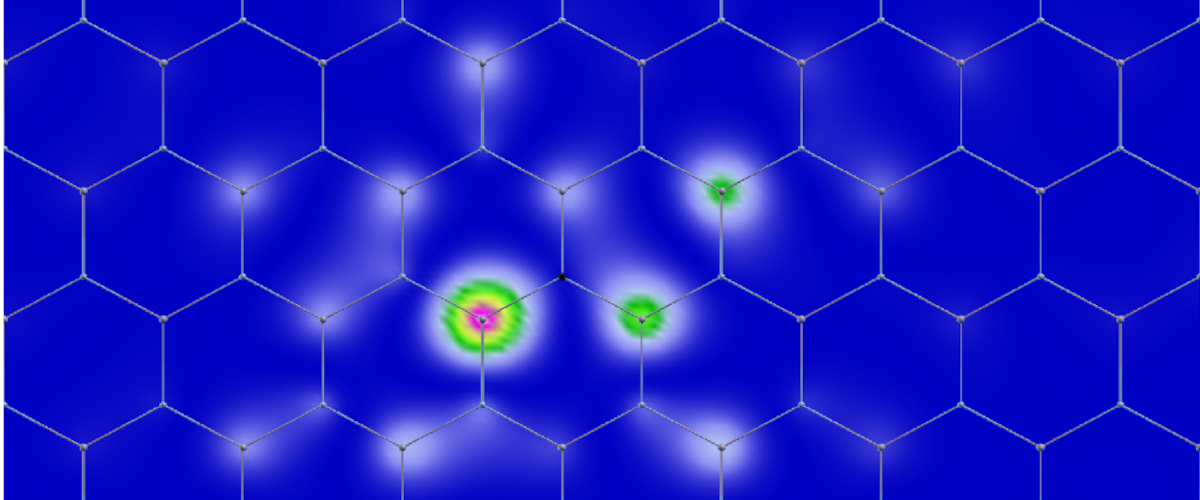
Exciton wavefunction imaging in h-BN. The black dot is the hole on N-atom (Phys. Rev. B, 99, 165201, (2019)).
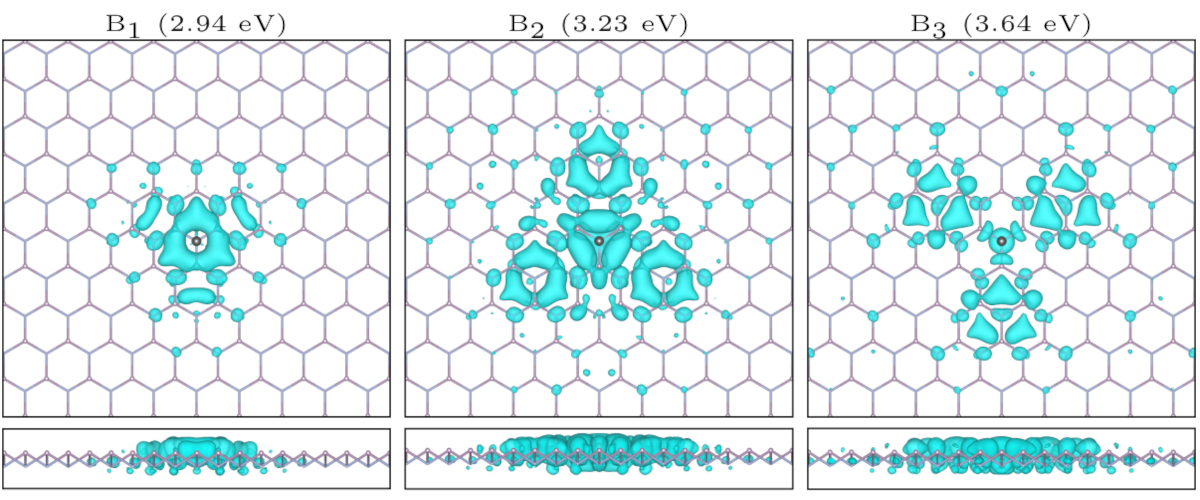
Most important bound excitons in NP (group V-V) monolayer (ACS J. Phys. Chem. C, 125, 12738, (2021).
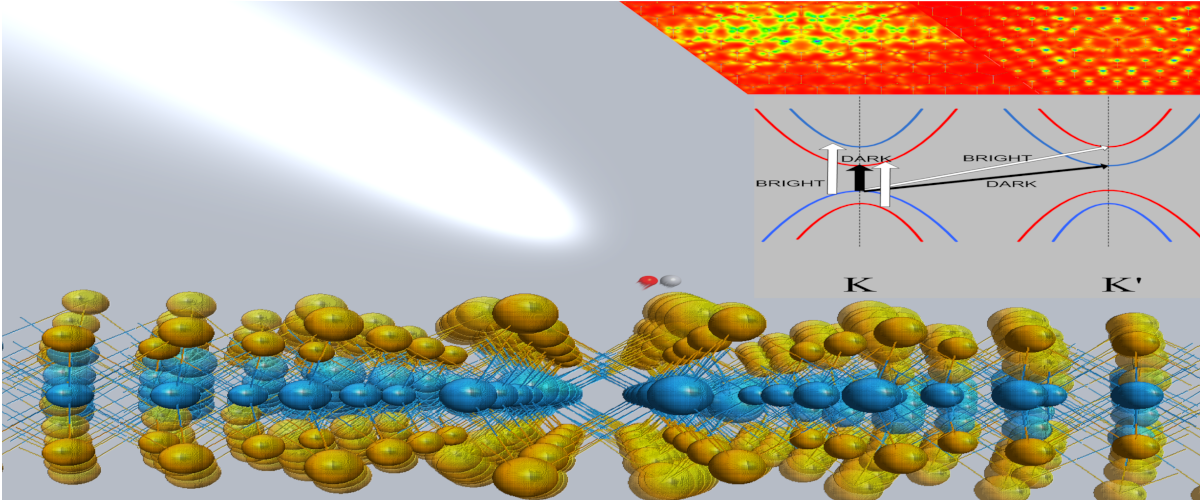
WSe2 exciton spin forbidden transition in presece of lattice vibrations (Phys. Rev. B, 98, 045143, (2018)).
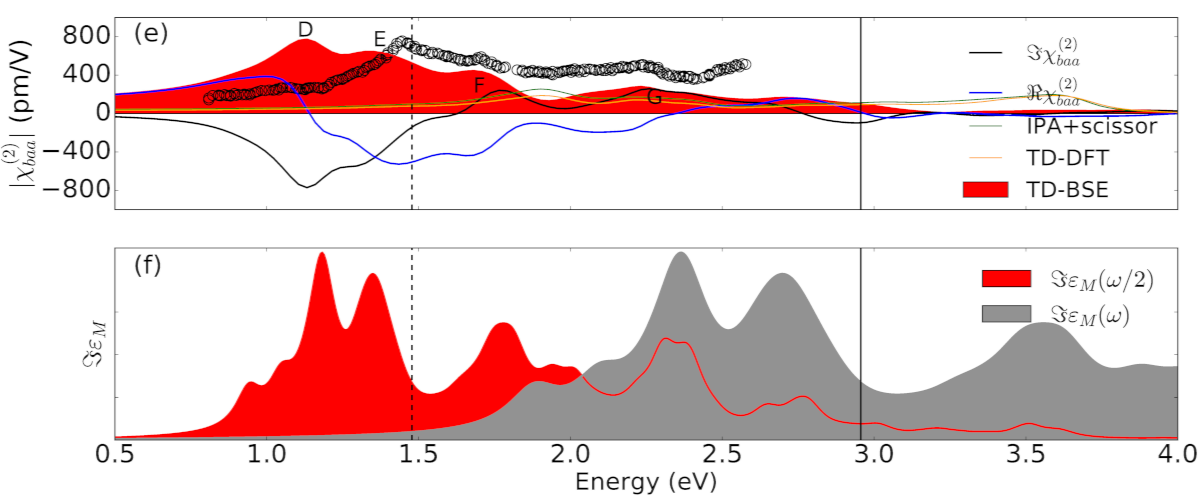
Second harmonic coefficient and 2ω resonances in GaAs monolayer. Symbols are from bulk GaAs (Phys. Rev. B 101, 155132, (2020)).
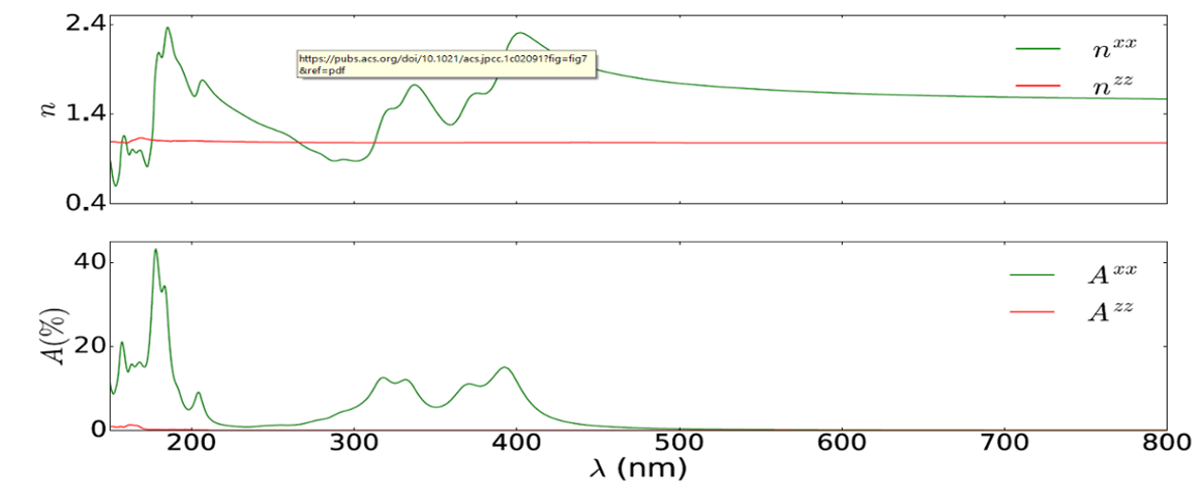
Refractive index and absorbance in NP (group V-V) monolayer from Bethe-Salpeter Equation (ACS J. Phys. Chem. C, 125, 12738, (2021))
Ab-initio/First principles calculations in two-dimensional materials
Electronic structure theory is the study of how electrons in a crystal interacts with atomic vibrations, light, defects and other external excitations via charges and energies that builds exciting electronic, thermal, magnetic and optical properties. In 1965, a series of two papers by Walter Kohn (Austrian born, American scientist) together with Pierre Hohenberg (French born American physicist) and Lu Jeu Sham (Chinese physicist) and later with John Pople (Bristish chemist) demonstrated that the their ground-state energy functional theory, the so-called “density functional theory” (sometimes also called as “ab-initio formalism” or “first principles theory), is inevitable to properly describe the electronic affairs inside crystal and attracted a Noble prize in chemistry (1998), shared between Kohn and Pople.
In early days, their theory was quite a challenge to solve because of lack of obvious computational resources, thanks to the now commercially available hardware accelerators and the high-speed data communicators, their equations are now both mathematically enjoyable to solve and develop.
In this group, we study the electronic correlations using the most advanced linear response many body perturbation theory. Such schemes are beyond the standard ground state energy functional theory and arrows to a more advanced Hartree-Fock theory, Random-phase approximation, self-consistent GW approximation and Bethe-Salpeter equation. These challenging schemes are computational-costly practice, but if mastered, can be done systematically and elegantly.